Wilms-Tumoren: Wie Gene und Prägung den Weg für Krebs ebnen
27.05.2025Ein Forschungsteam der Universität Würzburg hat neue Erkenntnisse zur Entstehung von Nierentumoren bei Kleinkindern gewonnen. Diese ermöglichen eine bessere Risikoeinschätzung und könnten die Grundlage für ein gezieltes Screening und eine verbesserte Früherkennung bilden.
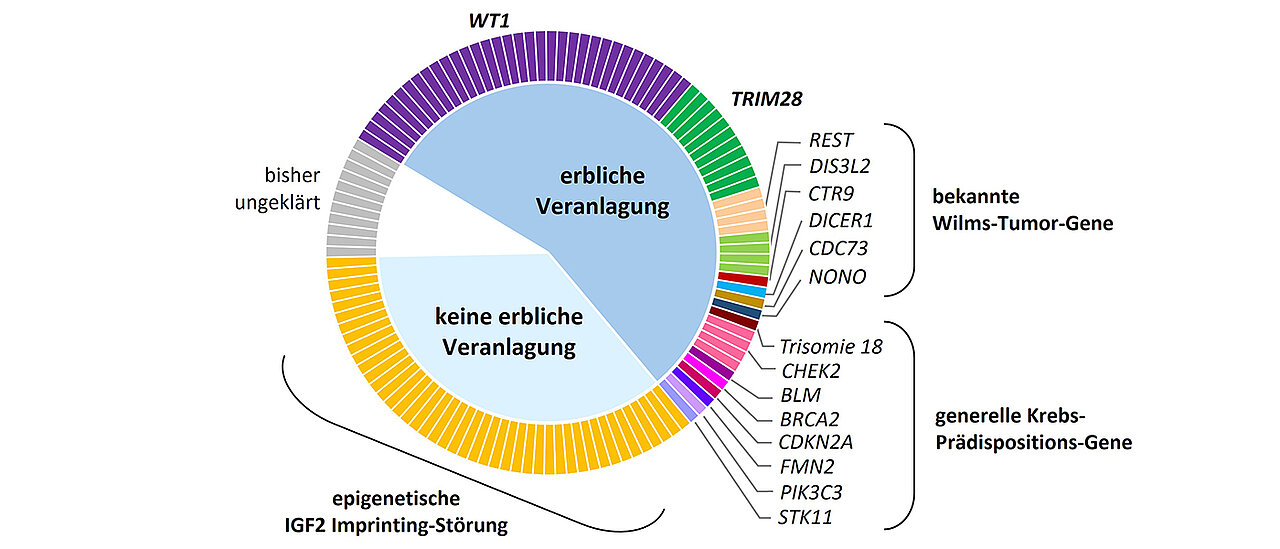
Ein Forschungsteam am Biozentrum der Julius-Maximilians-Universität Würzburg (JMU) hat zusammen mit Kooperationspartnern am Wellcome Sanger Institute in Cambridge (UK) einen bedeutenden Schritt zum Verständnis von Wilms-Tumoren, bösartigen Nierentumoren bei Kleinkindern, gemacht. Mithilfe der Proben der Wilms-Tumor-Biobank konnte das Team die erbliche Veranlagung (Prädisposition) für Wilms-Tumoren an einer großen Stichprobe systematisch entschlüsseln. Die Ergebnisse wurden jetzt in der Fachzeitschrift „Genome Medicine“ veröffentlicht; sie eröffnen neue Wege für die genetische Beratung und Überwachung von Risikopatienten.
Ein wissenschaftlicher Schatz: Die Wilms-Tumor-Biobank an der JMU
Die am Biozentrum der JMU angesiedelte Biobank für Wilms-Tumoren ist das Herzstück dieser Forschung. Im Zeitraum von knapp 30 Jahren (1994 bis 2022) haben die Verantwortlichen im Rahmen der deutschen Wilms-Tumor-Studie Proben von rund 1.800 betroffenen Kindern gesammelt. Unter diesen befanden sich 20 familiäre, also auch bei Eltern und/oder Geschwistern aufgetretene, Tumoren sowie 109 beidseitige (bilaterale) Tumoren, bei denen man von einer genetischen Prädisposition ausgeht.
„Bei über 90 Prozent dieser Fälle gelang es uns, die zugrundeliegende Veranlagung zu identifizieren“, erklärt Dr. Jenny Wegert, Mitarbeiterin am Lehrstuhl für Entwicklungsbiochemie und Erstautorin der Studie.
Schrittweise Tumorentstehung und stereotype Muster
Vor über 50 Jahren bereits postulierte Alfred Knudsen die sogenannte “Two-hit Hypothese“, welche erbliche Formen kindlicher Tumoren wie den Wilms-Tumor erklären sollte. Diese schrittweisen genetischen Veränderungen während der Tumorentstehung konnten die Forschenden jetzt in ihrer Studie im molekularen Detail nachweisen.
Am häufigsten fanden sie Mutationen in WT1, einem Tumorsuppressor-Gen, wobei zunächst eine der beiden Kopien des WT1-Gens in allen Körperzellen inaktiviert vorliegt. Dies allein kann schon mit einem erhöhten Risiko für Nierenschäden und bei Jungen mit Störungen der Geschlechtsentwicklung einhergehen.
Zur eigentlichen Tumorbildung kommt es jedoch erst, wenn auch die zweite Kopie des WT1-Gens in Nierenzellen ausfällt und gleichzeitig der Wachstumsfaktor IGF2 aktiviert wird, was zur Bildung von Tumorvorstufen führt. Ein letzter Schritt, die zusätzliche Aktivierung des WNT-Signalwegs, der viele Wachstums- und Differenzierungsprozesse steuert, ist dann für die Entwicklung des bösartigen Tumors verantwortlich.
Störungen der genomischen Prägung als Tumorauslöser
Für etwa die Hälfte der Patienten konnten die Wissenschaftlerinnen und Wissenschaftler genetische Veränderung in der Keimbahn und damit in allen Körperzellen als Ursache nachweisen. Neben WT1 waren davon auch zahlreiche weitere Gene betroffen, dies aber deutlich seltener.
„Ein überraschender Befund war jedoch, dass etwa ein Drittel der Kinder nicht eine der klassischen erblichen Mutationen aufwies, sondern eine Störung der sogenannten genomischen Prägung des IGF2-Gens“, sagt Jenny Wegert. Diese Prägung wird erst während der Embryonalentwicklung festgelegt und ist daher nicht vererbbar. „Das bedeutet, dass für Geschwisterkinder kein erhöhtes Risiko besteht und auch Betroffene die Tumorprädisposition nicht weitervererben“, so die Wissenschaftlerin.
Kinder mit dieser epigenetischen Prädisposition wiesen häufig „Mosaike“ auf, besaßen also nebeneinander Zellen mit normaler und Zellen mit gestörter IGF2-Prägung. Traten in Nierenzellen mit IGF2-Störung Mutationen in weiteren Genen auf, entwickelten sich Tumoren.
Konsequenz: Genetisches Screening für Risikopatienten
"Unsere neuen Erkenntnisse belegen eindrücklich, dass ein signifikanter Teil der kindlichen Nierentumoren eine erbliche Komponente hat", so Professor Manfred Gessler, Inhaber des Lehrstuhls für Entwicklungsbiochemie und Leiter der Studie. "Dies hat wichtige Folgen für die Klinik: In solchen Fällen besteht ein erhöhtes Risiko für Geschwister, und auch die Patienten selbst können später Zweittumoren entwickeln oder ein frühzeitiges Nierenversagen erleiden."
Die Studie spricht daher klar für eine breit angelegte molekulare Untersuchung von Blut- und Tumorproben der kleinen Patientinnen und Patienten. Ziel ist es, Fälle mit erhöhtem Risiko frühzeitig zu identifizieren und eine engmaschige Überwachung zu gewährleisten.
Originalpublikation
Wegert et al.: Distinct pathways for genetic and epigenetic predisposition in familial and bilateral Wilms tumor. Genome Medicine 17, 49 (2025).
https://doi.org/10.1186/s13073-025-01482-0
Kontakt
Prof. Dr. Manfred Gessler, Lehrstuhl für Entwicklungsbiochemie, T: +49 931 31-84159, manfred.gessler@uni-wuerzburg.de
Dr. Jenny Wegert, Lehrstuhl für Entwicklungsbiochemie, T: +49 931 31-81365,