Project A5 (terminated)
Introduction
Comparing proteomics and metabolomics allows new insights into S. aureus physiological growth. We update genome and proteome information and deliver flux models for three different S.aureus strains. We deal with the problem of limited data in metabolic modelling: All fluxes of the central metabolism in S. aureus are calculated according to selected external metabolite data applying a new calculation routine with fast convergence. This allows insights into the physiology of adaptation under nutrient and oxygen limitation. Furthermore, a time series of new 2D gel protein expression data published here is again used to estimate S. aureus fluxes. Comparison with the metabolite-predicted fluxes identifies strongly regulated enzymes for S.aureus under these physiological limitations. Finally, we analyze a bit more general the correlation between expression data and central metabolite fluxes in gram positive bacteria comparing S. aureus (above data sets) and Listeria monocytogenes (gene expression data sets versus isotopologue labelling data). A perspective section will summarize current projects extending this: Inclusion of internal metabolites, identification of enzyme complexes and metabolic switches on different levels.
Project A5 was successful, but as the research question was replaced by a new endeavor, it was terminated in 2009. For the research in the new project A8 (started in 2009) please go to its web-page.
Software developed for data analysis
| Comparative genomics study |
InGeno was developed for Comparative genomic studies. A detailed Staphylococci comparative study was performed using inGeno software. Genome of S. aureus COL is pairwise compared to each other Staphylococcus strains. Genes are marked by different colors, indicating the function categories.


InGeno has been published [1]: Liang and Dandekar, BMC Bioinformatics 2006,7:461.
The analysis input file for inGeno is available
| Comparison | data file |
|---|---|
| S. aureus COL vs S. aureus N315 | file1 |
| S. aureus COL vs S. aureus NCTC | file2 |
| S. aureus COL vs S. epidermidis ATCC | file3 |
Analysis snapshot files can be imported directly into inGeno software to acquire a rapid impression regarding gene comparison and functions. The original software and supplementary data for Staphylococcus aureus studies are available at here.
More information please read this webpage.
| Structure of transcription factors and functional genomics |
Virulence factors are investigated using GENOVA, which is a comprehensive editors allowing to modify, annotate the genome contexts including transcription factors. Researchers can also construct draft genomes using it, during our studies, we used it on investigating S. aureus RN1, RN1HG, Newman genomes. 
More information of GENOVA can be obtained in a publication [2] (Liang et al. Online Journal of Bioinformatics 2009, 10(2):201-19). More information can be obtained via this webpage.
| S. aureus RN1: file1 |
|---|
| Transcriptome data |
Dealing with incomplete genomes, JANE is able to map variable sequences (transcriptome data) to a reference genome with a specific alignment algorithm, thus achieve a rapid alignment/assembly. It provides an easy-to-use graphics interface for information retrieval and a toolkit fro EST or nucleotide sequence function prediction.
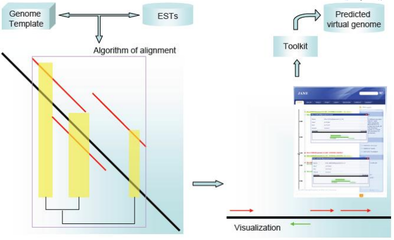
JANE has been published [3]: BMC Bioinformatics 2009,10:391. The algorithm has been implemented in a web server, publicly available at http://jane.bioapps.biozentrum.uni-wuerzburg.de.The demonstration data can directly acquired by clicking the button below the input-box.
A tutorial and sample files for JANE are available in the corresponding TR34webpage and the interface is available in the original website.
The analysis transcription file and reference genome sequences are also available: file1 (transcriptome data: JDK6008) and file2 (reference genome: Staphylococcus aureus COL).
| Metabolomics |
YANA is designed for metabolomic approaches. It allows to reconstruct the metabolic network in a genome scale. With this software, we have successfully built the model of S. aureus and completed the topological analysis and flux estimations [4].
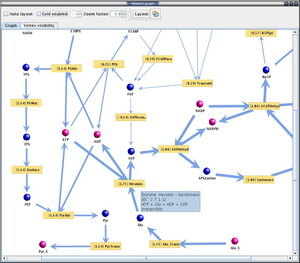
With YANA software, we are able to inspect the metabolic network of Staphylococci,
| model | file |
|---|---|
| Staphylococcus aureus N315 | model1 |
| Staphylococcus epidermidis | model2 |
| Staphylococcus saprophyticus | model3 |
| Towards Systems Biology |
YANAsquare evolves from YANA software. It offers a platform for rapid establishing the metabolic network of bacteria, such as aureus strains of interest. YANAsquare can utilize known reactions from online database, the tool has been integrated (known as KEGGBrowser). Possible flux modes of (EM/EP) can be calculated within the software. The scheme of SBML level-2 is compatible, components/compartments are generic supported. In addition to original YANA software, an optimized layout can be estimated by algorithms in this version. The method has been published in [5]: BMC Bioinformatics 2007, 8:313.
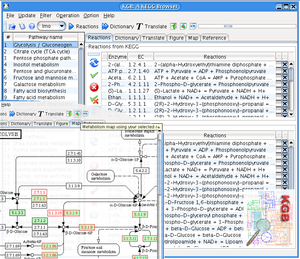
Using YANAsquare we established the metabolic mode for S. aureus strain. A sample file is available here (S. aureus COL)
References
[1] Liang C, Dandekar T: inGeno - an integrated genome and ortholog viewer fro improved genome to genome comparisons. BMC Bioinformatics 2006, 7:461.
[2] Liang C, Wolz C, Herbert S, Berhardt J, Engelmann S, Hecker M, Götz F, Dandekar T: GENOVA: A rapid genome visualization and functional genomics software applied to strain comparisons in Staphylococcus aureus. Online Joruan of Bioinformatics 2009, 10(2):201-219.
[3] Liang C, Schmid A, Lopez-Sanchez MJ, Moya A, Gross R, Bernhardt J, Dandekar T: JANE: efficient mapping of prokaryotic ESTs and variable length sequence reads on related template genomes. BMC Bioinformatics. 2009, 10:391.
[4] Schwarz R, Musch P, von Kamp A, Engels B, Schirmer H, Schuster S, Dandekar T: YANA - a software tool for analyzing flux modes, gene-expression and enzyme activities. BMC Bioinformatics 2005, 6:136.
[5] Schwarz R, Liang C, Kaleta C, Kühnel M, Hoffmann E, Kuznetsov S, Hecker M, Griffiths G, Schuster S, Dandekar T: Integrated network reconstruction visualization and analysis using YANAsquare. BMC Bioinformatics 2007, 8:313.